| Department of Pathology, State University of New York at Stony Brook |

 Go to: TOC |
|
|
|
This entity has also been called Hodgkin lymphoma, but several factors distinguish it from non-Hodgkin lymphoma. This remains true even though its lymphoid origin is increasingly established. |
|
|
Epidemiology
In the United States the incidence of HL in recent years has been 2.9 per 100,00, with whites more affected than blacks. In 1998, 7,100 new cases and 1,400 deaths are expected. Mostly because of increasingly effective therapy, the prognosis for HL is good. Currently versus 20-30 years ago, the death rate has declined about 60% and the five-year survival rate is 82% versus 40%. The age distribution in developed countries shows 2 modal peaks: young adults (15-34 y) and older adults (50+ y). In developing countries, the young adult peak is replaced by a childhood (0-14 y) form. This difference in pattern between developed and developing countries has suggested to some investigators that exposure to an infectious disease plays a role in HL's etiology. In fact epidemiology, serologic studies, and tissue probes all tend to associate many cases with Epstein-Barr virus. Nonetheless the etiologic role of the virus is controversial, and sensitive techniques are unable to find tissue evidence of the virus in half the cases.
Clinical
In parallel with this progression in subtype is a gradual spread from one lymph node region mostly to contiguous ones. HL originates usually within the neck or mediastinum, only to propagate across the diaphragm along the aortic and iliac lymph nodes and eventually to reach the spleen, liver, and bone marrow. Some cases display "B" or constitutional symptoms such as fever, night sweats and weight loss. Additional morbidity comes from the mass affect of the tumor and impaired cell mediated immunity. The outlook would be dismal (a 5 year untreated survival rate of 5%) were it not for effective chemotherapy and radiotherapy that achieve cures in up to 70% of cases. This intensely toxic treatment has its own dangers; second malignancies, most often acute myeloid leukemia, may crop up several years later. To minimize this danger, therapy is tailored to the extent of the disease as determined by a thorough staging. When its results would influence treatment, a staging laparotomy is performed, including splenectomy and biopsies of liver and retroperitoneal lymph nodes. The following staging criteria are used (and in fact may also be used for staging non-Hodgkin lymphoma): Ann Arbor Staging Classification

|
 ACTUALLY
I fibbed a while back when I said that the great white whale of hematopathology is T-cell lymphomas--it's really Hodgkin lymphoma(Hodgkin's disease and Hodgkin's lymphoma mean the same entity). The disease is named for Thomas Hodgkin, who first described the entity in 1832. For over a hundred years many a medical investigator's career shipwrecked while he or she struggled to harpoon the mystery of this ailment.
ACTUALLY
I fibbed a while back when I said that the great white whale of hematopathology is T-cell lymphomas--it's really Hodgkin lymphoma(Hodgkin's disease and Hodgkin's lymphoma mean the same entity). The disease is named for Thomas Hodgkin, who first described the entity in 1832. For over a hundred years many a medical investigator's career shipwrecked while he or she struggled to harpoon the mystery of this ailment.

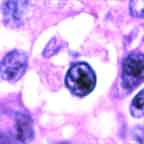
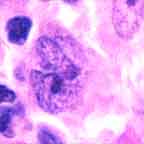
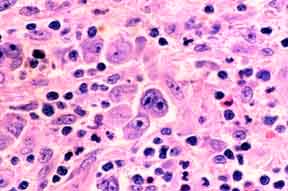 Note the presence both of diagnostic Reed-Sternberg cells and a suitable inflammatory background.
Note the presence both of diagnostic Reed-Sternberg cells and a suitable inflammatory background.
