| ชนิดของ Primary Immunodeficiency แบ่งตาม WHO Scientific group |
 |
| 4. Immunodeficiency with other abnormalities |
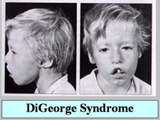
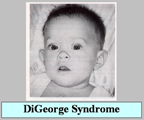
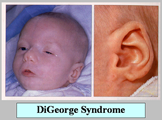
| 4.1 DiGeorge Syndrome (10) |
| เป็นโรคที่จัดอยู่ใน selective T-cell defects ประกอบด้วยความผิดปกติของ รูปหน้า อวัยวะต่างๆ (รูปที่ 8-10) และความผิดปกติทางภูมิคุ้มกัน |
|
กลุ่มอาการนี้ประกอบด้วย conotruncal cardiac malformation, hypocalcaemia, abnormal facial profiles และ immunodeficiency สาเหตุเกิดจากการพัฒนาที่ผิดปกติของ pharyngeal pouches ที่ 3 และ 4 (รูปที่ 11) ส่งผลให้มีความผิดปกติของการเกิด parathyroid glands และ thymus gland (รูปที่ 12) |
|
| ความผิดปกติทางหัวใจที่พบบ่อยในโรคนี้ คือ interrupted aortic arch, tetralogy of Fallot และ truncus arteriosus ความผิดปกติทาง chromosone ที่ก่อให้เกิดกลุ่มอาการนี้ คือ microdiletion ที่ chromosome 22q 11.2 สามารถใช้การตรวจด้วยวิธี FISH (Fluorescene In-situ Hybridization) เพื่อวินิจฉัยถึงความผิดปกตินี้ พบว่า 25% ของผู้ป่วยได้รับการถ่ายทอดความผิดปกติมาจากบิดามารดา ส่วนที่เหลือเป็นการเกิดขึ้นในตัวผู้ป่วยเอง |
| ภาวะ DiGeorge syndrome เป็นสาเหตุสำคัญของโรคหัวใจผิดปกติแต่กำเนิดอันเนื่องมาจากสาเหตุทางพันธุกรรม อย่างไรก็ดี ผู้ป่วยที่มีลักษณะการแสดงออกแบบเดียวกับกลุ่มอาการนี้อาจไม่พบความผิดปกติที่ chromosome ดังกล่าวก็ได้ |
| ความผิดปกติทางภูมิคุ้มกันมีได้หลายประการ ที่พบบ่อยได้แก่การมี CD3 (T-cell) ลดลง คือ ต่ำกว่า 1500 cells/mm3, CD4 ต่ำกว่า 1000 cells/mm3 และ มีภูมิต้านทานด้าน CMI ลดลง ส่วนมากความผิดปกติทางภูมิคุ้มกันจะดีขึ้นตามอายุ ผู้ป่วยบางรายอาจไม่มีความผิดปกติทางภูมิคุ้มกันเลยก็ได้ ไม่พบว่ามีลักษณะการแสดงออกของโรค (phenotype) ใด ที่สัมพันธ์หรือสามารถใช้พยากรณ์ได้ว่า ผู้ป่วยคนใดจะมีภูมิคุ้มกันบกพร่อง ในรายที่มีภูมิคุ้มกันบกพร่องอย่างมาก ซึ่งอาจเรียกว่า complete DiGeorge syndrome นั้น พบได้ประมาณ 5% ของผู้ป่วยกลุ่มนี้ อาจต้องทำ stem cell transplantation (SCT) หรือ Thymic transplantation เพื่อแก้ไขภาวะภูมิคุ้มกันที่บกพร่อง ผู้ป่วยที่สงสัยว่ามี DiGeorge syndrome แต่ไม่ทราบว่ามีภูมิคุ้มกันบกพร่องร่วมด้วยหรือไม่ ถ้าจำเป็นต้องได้รับเลือดต้องใช้ irradiated blood ไปก่อนจนกว่าจะทราบผลการตรวจทางภูมิคุ้มกัน เนื่องจากในเลือดที่ไม่ผ่านการฉายรังสีจะมี mature T-cell ซึ่งสามารถก่อให้เกิด graft versus host ได้ในผู้ป่วยที่มี T-cell deficiency |
| 4.2 Wiskott – Aldrich Syndrome (WAS)(11-12) |
| โรคนี้มีการถ่ายทอดทางพันธุกรรมแบบ X-linked ประกอบด้วยอาการ eczema, congenital thrombocytopenia และ recurrent infection โรคนี้เกิดจากการมี mutation ของ gene ที่ควบคุมการสร้าง WAS protein (WASP) โปรตีนนี้เป็น intracellular signaling molecule ที่มีความสำคัญในกระบวนการควบคุม actin cytoskeletal organization ของเซล นอกจากนี้ยังพบว่าโรค X-linked thrombocytopenia (XLT) ก็เกิดจาก mutation ของ gene นี้เช่นกัน แต่มีการแสดงออกเพียง thrombocytopenia โดยที่ไม่มีภาวะภูมิคุ้มกันบกพร่อง |
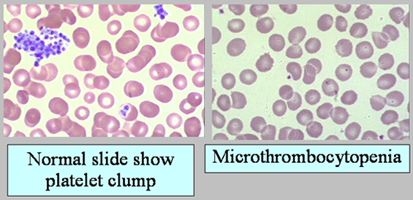
| ผู้ป่วยที่เป็น WAS เมือมาพบแพทย์ อาจไม่แสดงอาการที่กล่าวไว้ข้างต้นครบทุกอย่าง ทั้งนี้ขึ้นกับความรุนแรงของโรค ในระยะแรกบางรายอาจมีเพียง thrombocytopenia เท่านั้น โดยไม่มีอาการอื่นๆ หลังจากนั้นจึงมี eczema หรือ recurrent infection ตามมาเมื่อผู้ป่วยโตขึ้น อาการแสดงของโรคนี้ที่สำคัญ คือ thrombocytopenia ผู้ป่วยมักมาด้วย petechii และ bleeding tendency ตั้งแต่วัยทารก ถ้ารุนแรงมากอาจมีอาการตั้งแต่ neonatal period เกล็ดเลือดมักอยู่ระหว่าง 10,000 – 50,000 / mm3 แต่อาจสูงหรือต่ำกว่านี้ได้ อาการเลือดออกง่ายมีได้ในหลายระบบของร่างกาย เช่น อุจจาระมีเลือดปน บางครั้งผู้ป่วยได้รับการวินิจฉัยว่าเป็น infective diarrhea, intracranial hemorrhage และเลือดออกจาก umbilical stump หรือภายหลังการทำ circumcision สิ่งสนับสนุนการวินิจฉัยถ้าผู้ป่วย WAS มาพบในระยะนี้ แม้จะยังตรวจไม่พบ eczema หรือ recurrent infection ก็คือ การดูขนาดของเกล็ดเลือด อาจดูจาก blood smear หรือ platelet sizing โดยเครื่อง automated ผู้ป่วย WAS จะมีเกล็ดเลือดขนาดเล็ก (microthrombocytopenia) (รูปที่ 13) |
|
| โดยขนาดเฉลี่ยอยู่ที่ 3.8 fl ถึง 5.0 fl ค่าปกติ 7.0-10.5 fl ภาวะเกล็ดเลือดต่ำใน WAS มักจะไม่ตอบสนองต่อ steroid หรือ IVIG การทำ splenectomy อาจจะทำให้เกล็ดเลือดเพิ่มขึ้นได้ แต่เพิ่มความเสี่ยงต่อการเสียชีวิตจากการติดเชื้อจึงไม่แนะนำให้ทำ |
 Eczema พบได้ 80% ผู้ป่วยของ WAS แต่ไม่ใช่ลักษณะที่จำเป็นในการวินิจฉัยโรคนี้เพราะผู้ป่วย WAS บางคนอาจไม่มี eczema ส่วนใหญ่ eczema จะพบตั้งแต่ก่อนอายุ 6 เดือน (รูปที่ 14) การติดเชื้อซ้ำๆ มักเป็น sinopulmonary infection และการติดเชื้อที่ต้องอาศัย T-cell ในการกำจัด เช่น Pneumocystis carinii pneumonia และ recurrent herpes infection ก็พบได้ในผู้ป่วยโรคนี้ Eczema พบได้ 80% ผู้ป่วยของ WAS แต่ไม่ใช่ลักษณะที่จำเป็นในการวินิจฉัยโรคนี้เพราะผู้ป่วย WAS บางคนอาจไม่มี eczema ส่วนใหญ่ eczema จะพบตั้งแต่ก่อนอายุ 6 เดือน (รูปที่ 14) การติดเชื้อซ้ำๆ มักเป็น sinopulmonary infection และการติดเชื้อที่ต้องอาศัย T-cell ในการกำจัด เช่น Pneumocystis carinii pneumonia และ recurrent herpes infection ก็พบได้ในผู้ป่วยโรคนี้
การตรวจทางภูมิคุ้มกันในโรคนี้จะพบว่า มี IgM ต่ำ ส่วน IgG มักปกติ มีการสร้าง antibody ต่อ pneumococcal polysaccharide ผิดปกติ และระดับ isohemagglutinin ต่ำ คนไข้ส่วนใหญ่จะมีการทำงานของ T-cell ลดลง การรักษาโรคนี้คือการทำ SCT ซึ่งจะแก้ไขทั้งความผิดปกติทางภูมิคุ้มกันและ ภาวะ thrombocytopenia ตลอดจน ภาวะแทรกซ้อนอื่น ๆ เช่น autoimmune และ malignancy SCT จะได้ผลดีถ้าทำตั้งแต่อายุน้อยกว่า 5 ขวบ
ผู้ป่วย WAS ที่ไม่ได้รับการรักษาโดยทำ SCT จะมีโอกาสเสี่ยงต่อการเกิดมะเร็งบางชนิดมากกว่าคนปกติ เช่น leukemia, lymphoma และ EBV-associated tumors นอกจากนี้ยังมีโอกาสเกิด autoimmune disease เช่น hemolytic anemia, arthritis, vasculitis, inflammatory bowel disease และ glomerulonephritis มากกว่าคนปกติ สาเหตุการตายในผู้ป่วย WAS ที่ไม่ได้รับการทำ SCT นั้น 40-50% เกิดจากการติดเชื้อ, 25% เกิดจากภาวะเลือดออกผิดปกติ และ 25% เกิดจาก malignancy |
| 4.3 Ataxia – Telangiectasia (AT)(13-14) |
| โรคนี้ถ่ายทอดแบบ autosomal recessive อาการของโรค ได้แก่ cerebellar ataxia, oculocutaneous telangiectasia และมี combined immunodeficiency ผู้ป่วยจะมีความเสี่ยงต่อการเกิดมะเร็งมากกว่าคนปกติ โรคนี้พบว่ามี mutation ของ gene ซึ่งเรียกว่า ATM (AT mutated) gene ซึ่งเกี่ยวข้องกับ cell cycle control และ การตอบสนองของ cell ต่อ DNA damage เมื่อกลไกนี้ผิดปกติจึงทำให้ cell กลายเป็นเนื้อร้ายได้ง่าย อาการนำของโรคนี้ คือ อาการทางระบบประสาท ซึ่งมักแสดงออกโดยความผิดปกติของการเดิน (ataxic gait) ซึ่งเกิดขึ้นในขวบปีที่สองของชีวิต อาการ ataxia จะเป็นมากขึ้นและลามจาก trunk, extremities ไปที่ palatal muscles ทำให้เกิดการพูดที่ผิดปกติ (dysarthric speech, drooling, choreoathetosis) จนในที่สุดผู้ป่วยจะไปไหนมาไหนเองไม่ได้เมื่ออายุราว 10 ปี ส่วน telangiectasia พบที่นัยน์ตาขาว (sclera) และผิวหนัง จะเกิดเมื่ออายุประมาณ 4-8 ปี ความผิดปกติทางภูมิต้านทานมีความรุนแรงแตกต่างกันในผู้ป่วยแต่ละราย แต่มักทำให้มีการติดเชื้อทางเดินหายใจซ้ำ ๆ และอาจทำให้เกิด chronic lung disease การติดเชื้อนี้มักชัดเจนเมื่ออายุ 3-6 ปี การให้ IVIG ทำให้การติดเชื้อลดลงได้ โรคทางเดินหายใจและ aspiration pneumonia เป็นสาเหตุการตายอันดับแรกของผู้ป่วย AT รองลงมา ได้แก่ malignancy ซึ่งมักเป็น T-cell หรือ B-cell malignancy เช่น leukemia และ lymphoma ผู้ป่วยโรคนี้จะมีระดับ alpha fetoprotien (AFP) และ carcinoembryonic antigen (CEA) สูงขึ้นในเลือด ซึ่งสามารถใช้ช่วยในการวินิจฉัยได้ |
| ความผิดปกติทางภูมิคุ้มกันในโรคนี้เป็นชนิด combined defects โดยมีปริมาณ T และ B-cell ลดลง อาจมี reverse CD4: CD8 ratio และอาจมีการลดลงของ in vitro T-cell proliferation ระดับ IgA จะลดลง ในผู้ป่วยบางรายอาจจะมี IgG2 และ isohemagglutinin ลดลงด้วย |
   |